
| Livestock Research for Rural Development 25 (3) 2013 | Guide for preparation of papers | LRRD Newsletter | Citation of this paper |
The objective of this study was: 1) to evaluate a two-step selection strategy involving genome-wide selection (GS) versus phenotypic and pedigree-based selection methods, and 2) to compare the differential returns from selection schemes in a nucleus swine breeding program. Annual genetic gain and return per dollar invested were used as evaluation criteria. Data for the study was generated by simulation of relevant target populations typical of swine population structure in developing countries and assuming that genetic improvement was concentrated on purebreds in a nucleus breeding program. Selection of replacement candidates were based on selection indices constructed for all traits in the breeding objectives for pig production in developing countries. Traits in the selection index included number born alive, feed conversion ratio, loin eye area and average daily gain.
The result showed that accuracy of genomic indices for the pre-selection and selection strategy ranged from 0.38 to 0.66 and were greater than the accuracy of conventional selection indices (0.10 – 0.64). Genome-wide selection generated an increase of about 38% to 173% in annual returns compared to other conventional approaches for a population in linkage disequilibrium. The expected return per dollar invested in a GS scheme was comparable to conventional methods when the cost of genotyping for 60 K single nucleotide polymorphism panel was at $30 per sample. This genotyping cost is realisable using genotype imputation technique and as costs for denser marker panel will likely decrease in the near future.
Key words: differential returns, developing countries, genome-wide selection, simulation
Nucleus breeding schemes (NBS) describe a breeding program in which selection is carried out on a small number of elite breeding animals in a nucleus that produces breeding stock for use in a multiplier or commercial population. The NBS has been proposed to overcome the technical constraints limiting the implementation of genetic improvement of pigs for low-input production systems in developing countries (Smith 1988; Hodges 1990). The genetic superiority generated in the nucleus is disseminated to the whole population through breeding. Genetic evaluation to select replacement stock is implemented after a performance testing program has been completed for all possible candidates (de Roo 1987).
Performance testing is utilized to prevent biases in estimated breeding value (EBV) caused by preferential treatment (Kuhn et al 1994) and to avoid the impact of heterogeneous variances in different environments (Garrick and van Vleck 1987). The performance testing of young boars and gilts is associated with a substantial cost, usually an average of $55 per tested pig based on Canadian prices (B. Sullivan, personal communication, 2011). This price is based on costs for animal registration, performance testing, cost for marketing the non-selected intact males, costs for raising purebred females and cost for genetic evaluation. Owing to the cost of performance testing, swine breeders often pre-select the animals to be tested. In developing countries, pre-selection of candidates for performance testing is based on knowledge of the phenotypic performance of their parents and when they have attained certain age or body weight (Dzama 2002). In addition, selection of replacement stock from tested candidates is strictly on the pigs own performance excluding records of its relatives (Aker and Kennedy 1988; Dzama 2002).
With the advent of genome-wide selection (GS) based on genomic breeding values (GBV) predicted from dense single nucleotide polymorphism (SNP) marker genotyping (Meuwissen 2001; Muir 2007), a genomic index (Dekkers 2007) of traits in the breeding objective can be constructed for all possible candidates and used for pre-selection of individuals entering the test station and for selection of replacement candidates. The objective of this simulation study was to evaluate a two-step selection strategy involving genome-wide selection versus phenotypic and pedigree-based selection methods, and to compare the differential returns from selection schemes in a nucleus swine breeding program. Two scenarios involving previously selected populations in high linkage disequilibrium (LD) and unselected populations in low LD were studied.
All pigs found in developing countries were assumed to come from a common base of unrelated individuals which served as ancestors to the selected and unselected populations. The common base population was mated based on the union of gametes randomly sampled from the male and female gametic pools for 1000 generations with effective population size (Ne) of 5000 breeding individuals. The ancestral population was contracted to Ne of 1050 individuals consisting of 300 breeding males at the end of another 1000 generations in order to generate initial historic LD. At generation 2001, the ancestral population was split into two unequal sized purebred subpopulations referred to as selected and unselected populations in this study. The selected population was created by drawing the top ranking 50 males and top ranking 250 females based on their phenotypes from the common base population resulting in Ne of 167 and were subjected to phenotypic selection for the trait under consideration for 50 generations with an intensity of 10% for males and 50% for females. Each female produced 4 offspring (2 males and 2 females) resulting in 1000 breeding animals in each generation from which selection was made. The remaining members of the common base population consisting of 250 males and 500 females were allowed to mate at random concurrently for 50 generations resulting in the unselected population with Ne of 667. The Ne assumed in this study was chosen to yield a simulated level of LD observed in real life for selected exotic pigs (Du et al 2007) and unselected indigenous pigs (Amaral et al 2008).
At generation 2050, two purebred lines that consisted of a previously selected population in high LD between adjacent markers (r2 = 0.29) and unselected population in low LD (r2 = 0.05) were produced and served as the source of foundation stock for the nucleus and for the two scenarios investigated. A 250 sow operation that is capable of producing on average two litters per sow per year was assumed. A total of 250 sows and 25 boars were randomly chosen from each of the purebred lines to establish the foundation stock for the nucleus. These were randomly mated to produce 20 piglets per sow per year resulting in a total of 5000 piglets annually assuming litter size and litter rate are already corrected for fertility and piglet survival. Besides the nucleus population, another population was simulated for each trait starting from the same base population as in the nucleus. This population was used as a training set to estimate the marker effects to be used as priors for the application of GS in the nucleus breeding scheme. This design eliminates the effect of close relationship between the training set and the prediction set in the nucleus.
The simulated genome consisted of 5 chromosomes of 150 cM each with 250 multi-allelic segregating quantitative trait loci (QTL) and a marker density of 22 bi-allelic loci per cM evenly spaced across the genome to represent approximately 60 K SNP marker panel that is currently available (Ramos et al 2009). The Haldane mapping function with no interference and a Mendelian inheritance of all loci was assumed. The allelic effects of the QTL including original and new mutations were sampled from a gamma distribution with a shape parameter of (Hayes and Goddard 2001) and scaled such that the sum of the QTL variances in the last historic generation equals the input QTL variance. The markers and QTL were simulated with equal starting allele frequencies and a recurrent mutation rate of 2.5 x 10-3 for markers and infinite-allele mutation rate of 2.5 x 10-5 for QTL.
The traits included in the breeding objective were number born alive (NBA), feed conversion ratio (FCR), loin eye area (LEA) and average daily gain (ADG). The genetic parameters used for simulating the data are given in Table 1. Traits were simulated independently assuming a finite additive locus model such that the true breeding value of an individual was equal to the sum of the 250 QTL effects scaled on the basis of the specified heritability and phenotypic variance. No pleiotropy or genotype-environment interaction was simulated. The genetic parameter estimates used for simulation were from a meta-analysis study carried out for pig production traits in tropical developing countries (Akanno et al 2012). However, the economic values for each trait were based on Canadian prices obtained from Canadian Centre for Swine Improvement (CCSI) website. All cost figures are presented in Canadian dollars.
|
Table 1. Traits in the breeding objective and their parameters |
||||
|
Variables |
NBA |
ADG |
FCR |
LEA |
|
Heritability |
0.08 |
0.28 |
0.32 |
0.51 |
|
Standard deviation |
2.78 |
101.79 |
0.35 |
3.85 |
|
Economic value, $ |
18.07 |
3.02 |
-5.17 |
1.13 |
|
NBA = number born alive; ADG = average daily gain; FCR = feed conversion ratio; LEA = loin eye area |
||||
In this era of genomics, it is reasonable to assume the availability of a repository of marker effect estimates defined for specific locations on a given SNP panel and for a given population. Using the same SNP panel, individuals can be genotyped at any time and their GBVs obtained for selection decision based on prior estimates of marker effects from a similar training population. The assumption is that marker density is high enough to capture useful LD between markers and QTL leading to high accuracy of selection. This is akin to the conventional method where genetic parameter estimates from other sources are used as priors in genetic evaluation of a novel similar population in order to obtain EBVs for selection purposes.
In this study, the population designated as the training set was also evaluated using a pedigree-based best linear unbiased prediction (BLUP) model for comparison. Comparisons were based on accuracy between true and estimated breeding values for the training and validation populations. The training set consisted of 2275 individuals and 15100 segregating markers with minor allele frequency (MAF) ≥ 0.10, while the validation set consisted of 5000 breeding candidates produced annually in the nucleus.
The pedigree-based BLUP model assumed was according to (Henderson 1984). The model used was
y = μ1n + Za + e 1
where y is a vector of observed phenotype of size n, μ is the overall mean, 1n is a vector of n ones, a is a vector of random additive genetic effects of each individual ~N (0, Aσ2a), A is the additive relationship matrix, Z is a design matrix for the additive genetic effects and e is a vector of random residuals ~N (0, Iσ2e). The variance ratio required in the mixed model equations was (1-h2)/h2. The ASREML program (Gilmour et al 2009) was applied for estimation of genetic effects and variances.
For the genome-based model, the marker effects were estimated using ridge regression described in Meuwissen et al (2001) and extended by Fernando et al (2007). The statistical model was
y = μ1n + Wg + e 2
The mixed model equations were:
where y is a vector of observed phenotype of
size n, g is a vector of SNP allele substitution effects ~N
(0, I
![]()
![]() ),
W is a design matrix that contains 0, 1 or 2 for the number of one of the
two alleles in the SNP genotypes,
),
W is a design matrix that contains 0, 1 or 2 for the number of one of the
two alleles in the SNP genotypes, ![]()
![]() is
the average base SNP heterozygosity, σ2g is the
genetic variance estimated using ASREML program and m is the total number
of markers. The other parameters are as described before.
is
the average base SNP heterozygosity, σ2g is the
genetic variance estimated using ASREML program and m is the total number
of markers. The other parameters are as described before.
Once estimates of marker effects were obtained from the training set, the GBVk of kth animal in the training and for the future candidates in the nucleus (prediction set) was computed as:

All individuals in the rearing piggery were pre-selected to enter into the test station for performance testing (Figure 1). Three pre-selection strategies were applied namely: 1) phenotypic selection which uses a phenotypic index (PI) constructed from parent average phenotypic performance of all individual’s in the rearing piggery as follows:

where vl is the economic
weight of trait l, Pl is an individual’s parent average
of phenotypic performance for trait l, QUOTE
![]()
![]() is
the mean parent average of phenotypic performance for trait l,
is
the mean parent average of phenotypic performance for trait l,
![]()
![]() is
the trait heritability and t is the number of traits in the index, 2)
pedigree selection based on selection index (SI) calculated for each individual
as follows:
is
the trait heritability and t is the number of traits in the index, 2)
pedigree selection based on selection index (SI) calculated for each individual
as follows:

where vl is the economic weight of trait l, PAl is the predicted parent average breeding values of an individual for trait l obtained by using the BLUP model in equation 1 and t is the number of traits in the index, and 3) genome-wide selection based on a genomic index (GI) constructed for all individuals in the rearing piggery as follows:

where vl is the economic weight of trait l, GBVl is the genomic breeding values of an individual for trait l predicted using equation 4 assuming that estimates of marker effects were available from a similar training population and t is the number of traits in the index. All individuals in the rearing piggery were assumed to be genotyped for the same SNPs as in the training population. A total of 1000 top ranking individuals made up of male and female candidates were pre-selected for performance testing based on the selection indices described above.
 |
|
Figure 1. Decision point in a nucleus swine breeding program |
In order to predict genetic gain per year for the whole operation to compare between selection schemes implemented, records were assumed to be available for all performance tested candidates at the end of the test and for all traits studied. Three selection criteria were applied namely; 1) phenotypic index based on an individual’s own phenotype, 2) selection index based on BLUP evaluation of animals with observation and 3) genomic index based on marker effects re-estimated for all traits using phenotype from performance testing. The accuracy of selection was determined as correlation between the index and the aggregate genotype (H: the product of the economic value and true breeding value for each trait summed across all traits in the breeding objective) for all tested candidates.
In another scenario for the genome-wide selection scheme, accuracy of GI for pre-selection strategy was applied to predict genetic gain per year. This allowed for the evaluation of a strategy that completely eliminates performance testing program assuming that marker effects will continue to be available from other sources.
Assuming that 5% of the young boars and 50% of gilts were selected annually as replacement stock from all tested candidates, expected genetic gain per pig per year was predicted as follow:

where i is the selection intensity derived based on the proportion of each sex selected, rHI is the correlation between the aggregate genotype and the selection index in males and females, L is the generation interval which was assumed as 1.8 years based on Canadian average and σH is the standard deviation of the aggregate genotype which was derived by solving equation 9 using parameters from Table 1 and assuming that covariance between traits was zero:

where v is the economic weight and 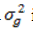 is
the genetic variance of traits in the aggregate genotype. The expected standard
deviation of the aggregate genotype (σH) worked out to be
$178.95. Return per year was calculated by multiplying
is
the genetic variance of traits in the aggregate genotype. The expected standard
deviation of the aggregate genotype (σH) worked out to be
$178.95. Return per year was calculated by multiplying
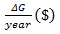 by
5000 assuming that the nucleus produces 5000 improved breeding stocks annually.
Depending on the scenario evaluated, total cost involved in the operation
included cost of genotyping 5000 breeding candidates for pre-selection and the
cost of performance testing 1000 pre-selected candidates. The return on
investment was obtained as total returns minus total cost while return per
dollar invested was obtained by dividing return on investment by total cost.
All analyses were coded in R statistical software (Ihaka and Gentleman 1996) and
each scenario was replicated ten times.
by
5000 assuming that the nucleus produces 5000 improved breeding stocks annually.
Depending on the scenario evaluated, total cost involved in the operation
included cost of genotyping 5000 breeding candidates for pre-selection and the
cost of performance testing 1000 pre-selected candidates. The return on
investment was obtained as total returns minus total cost while return per
dollar invested was obtained by dividing return on investment by total cost.
All analyses were coded in R statistical software (Ihaka and Gentleman 1996) and
each scenario was replicated ten times.
The accuracy of EBVs in the training and validation sets when using the two selection methods (pedigree and genomics) and for traits in the breeding objective is given in Table 2. Accuracy tended to increase with increasing trait heritability across methods and populations. Genome-wide selection had greater accuracy than pedigree-based method in the training and validation sets and for all traits in the breeding objective. The validation result for the pedigree method was based on parent average of individuals in the nucleus. Previously selected population in high LD had lower accuracy than unselected population in low LD for different traits and selection methods due to differences in selection pressure which reduced the genetic variation in the selected population (Bulmer 1976) and also due to difficulty in the estimation of marker allele substitution effects at low MAF in the selected population. In addition, GS method had greater accuracy than the pedigree method because GS acts directly on the QTL in LD with markers to improve accuracy (Habier et al 2007). The accuracies of genetic evaluation given in this study for different traits were in agreement with results from previous simulation studies at similar trait heritability (Meuwissen et al 2001; Muir 2007).
|
Table 2. Accuracy1 of estimated breeding values of pigs in the training and validation sets for traits in the selection index |
||||
|
Populations |
Traits |
Methods |
Training set |
Validation set2 |
|
Selected |
NBA |
Pedigree |
0.36±0.01 |
0.10±0.02 |
|
|
|
Genomics |
0.49±0.02 |
0.43±0.03 |
|
|
ADG |
Pedigree |
0.41±0.02 |
0.15±0.02 |
|
|
|
Genomics |
0.54±0.02 |
0.47±0.02 |
|
|
FCR |
Pedigree |
0.40±0.02 |
0.13±0.02 |
|
|
|
Genomics |
0.51±0.02 |
0.46±0.02 |
|
|
LEA |
Pedigree |
0.45±0.02 |
0.19±0.02 |
|
|
|
Genomics |
0.58±0.02 |
0.52±0.03 |
|
Unselected |
NBA |
Pedigree |
0.47±0.02 |
0.19±0.02 |
|
|
|
Genomics |
0.55±0.02 |
0.44±0.01 |
|
|
ADG |
Pedigree |
0.69±0.01 |
0.40±0.02 |
|
|
|
Genomics |
0.77±0.01 |
0.67±0.02 |
|
|
FCR |
Pedigree |
0.70±0.01 |
0.42±0.02 |
|
|
|
Genomics |
0.78±0.01 |
0.68±0.02 |
|
|
LEA |
Pedigree |
0.80±0.01 |
0.50±0.02 |
|
|
|
Genomics |
0.85±0.00 |
0.74±0.01 |
|
1Correlation between true and estimated breeding values averaged over 10 replicates 2Validation result for pedigree method is based on parent average of individuals in the nucleus |
||||
Table 3 shows the correlation (rHI) between aggregate genotype and various selection indices applied for pre-selection and selection strategies. Accuracy of genomic index (GI) for pre-selection strategy was similar to the average accuracy based on a single trait (Table 2). For a GS scheme, the accuracy of the GI is expected to be slightly lower than the accuracy based on a single trait (Sitzenstock et al 2010) because of the weighting effect of the economic values on the trait breeding values. Similar trend was observed when comparing accuracies of selection index to the average accuracy for a single trait in the pedigree-based method. The accuracy of GI in the selected population was slightly lower than the values observed for unselected population due to differences in selection pressure. For unselected population, accuracy of GI was similar to the values reported by König and Swalve (2009) and Sitzenstock et al (2010) in simulation studies.
For pre-selection strategy, accuracy of GI used was greater than the accuracy of conventional methods in the selected and unselected populations (Table 3). The pre-selection of candidates using the conventional approach had lower accuracy because they were constructed based on parent average. In real life, candidates for pre-selection have no records of their own to allow for construction of selection or phenotypic indices. Swine breeders in developing countries usually pre-select candidate for performance testing based on knowledge of their parents performance (Dzama 2002). For example, a breeder will pre-select a candidate for performance test if it comes from a sow with greater than 10 piglets in a litter or gilt with greater than 7 piglets in a litter. The availability of genomic information early in life of selection candidates offers the opportunity to make selection decision that is accurately informed on the basis of an individual’s genetic merit.
Furthermore, accuracy of genomic index for pre-selection strategy was greater than the accuracy of phenotypic and BLUP selection indices when records became available in the nucleus for genetic evaluation (Table 3). This is because GS uses dense marker panel to exploit LD between marker and QTLs affecting traits in the index leading to greater accuracy of selection. When marker allele substitution effects were re-estimated for all traits using the record of performance of 1000 breeding candidates from the test station, accuracy of GI was greater than the accuracy of conventional selection indices in the selected population but slightly lower than accuracy of BLUP selection index in unselected population. This decrease in rHI value in the unselected population when marker effects were re-estimated was due to the sample size used in the re-estimation (1000 vs. 2275 individuals used previously for training) such that QTL allele effects were likely estimated with lower accuracy due to lower average MAF. Accuracy of GS is expected to increase when re-estimating marker allele effects from a large reference dataset of greater than 2000 individuals (Meuwissen et al 2001).
|
Table 3. Accuracy of selection indices (rHI) for two steps selection strategies using different methods and for different populations in a nucleus swine breeding program |
||||
|
Populations |
Methods |
Group |
Pre-selection |
Selection |
|
Selected |
Phenotypic |
Males |
0.10±0.01 |
0.17±0.01 |
|
|
|
Females |
0.11±0.01 |
0.16±0.01 |
|
|
Pedigree |
Males |
0.12±0.02 |
0.33±0.05 |
|
|
|
Females |
0.13±0.02 |
0.33±0.04 |
|
|
Genomics |
Males |
0.45±0.02 |
0.39±0.03 |
|
|
|
Females |
0.47±0.02 |
0.38±0.03 |
|
Unselected |
Phenotypic |
Males |
0.33±0.02 |
0.44±0.02 |
|
|
|
Females |
0.32±0.01 |
0.42±0.03 |
|
|
Pedigree |
Males |
0.36±0.02 |
0.64±0.02 |
|
|
|
Females |
0.36±0.02 |
0.64±0.02 |
|
|
Genomics |
Males |
0.65±0.02 |
0.62±0.02 |
|
|
|
Females |
0.66±0.02 |
0.62±0.02 |
The expected accuracy of phenotypic index currently employed in nucleus test stations in developing countries was lower than the accuracy for conventional BLUP and genome-wide selection methods and was greatly reduced in the selected population than in the unselected population. For all methods of selection, the accuracy of selection indices was similar in males and females because neither the young boars nor the gilts have progeny with records. When parent records were included in the evaluation, males had greater accuracies than females due to more information on the sire pathway (result not shown). Under a GS scheme, genetic evaluation can proceed on selection candidates based on their own genotype and phenotype information without requiring knowledge of their parent records (Meuwissen et al 2001) which is not the case for BLUP.
As in developed countries, non-successful attempts have been made over the past two decades to institute a pedigree recording system that will allow traditional selection strategies for swine improvement in developing countries (Kahi et al 2005), NBS was proposed as an alternative by selecting within a nucleus using conventional approach (Smith 1988). Performance testing of individuals in the nucleus was an additional strategy to compare the genetic merit of potential candidates for selection of replacement stock. Under a performance testing program, the use of an individual's own performance as a measure of its genetic merit is reliable for traits of high heritability like loin eye area and average daily gain. An individual’s estimated genetic merit can further be improved by including the records of its relatives in the genetic evaluation program. However, it becomes problematic when dissecting an animal genetic merit for a trait with low heritability like number born alive. More records would need to be collected in order to accurately estimate breeding value for such a trait. In addition, developing a pedigree recording system for implementing a conventional BLUP method can be very costly and may be impracticable for developing countries, genome-wide selection can supplant the development of this infrastructure.
Our results shows that the availability of genetic markers will help to improve the accuracy of selection for traits with low heritability and will provide GBV early in life of selection candidates which offers an opportunity to reduce the cost of performance testing. In real life more records will need to be collected from performance tested candidates and added to the records from training set for re-estimation of marker effects in order to improve accuracy of selection. As an alternative, GS can be implemented based on the accuracy of pre-selection strategy assuming that estimates of marker effects will continue to be available from other sources which eliminates completely the practice of performance testing by keeping only individuals with GI that are above the threshold for selection. The selected candidates are then raised to reproductive maturity and can be mated early in life, thus reducing both generation interval and the cost of housing and performance testing. In addition, by preselecting individuals that enter the test station, potential candidates can further be evaluated for secondary traits such as conformation and health. However, will farmers accept genomic indices as a selection criterion without having a prior knowledge of the performance of selection candidates in a test station? This will be the crucial point for practical implementation of GS in a nucleus swine breeding program in future.
The expected genetic gain per pig per year in the nucleus is instrumental in showing the differences between the various selection methods employed (Table 4). For the selected population in high LD, using GS based on re-estimated marker effects to select replacement stock will achieve about 132% and 17% greater returns per year compared to current phenotypic selection method and a possible pedigree-based approach, respectively. The expected increase in returns of 17% was less than the 37% increase in returns reported by Simianer (2009) using a deterministic approach to evaluate GS. When GS was used to replace phenotypic selection in an unselected population in low LD, annual returns was increased by 43% but was reduced by 3% when substituting for a pedigree method. This outcome supports the fact that having sufficient level of LD in the population of interest is central to the functionality of GS. For population with low LD, a greater marker density than 60 K SNP panel currently employed in this study may lead to more accurate GBV.
Under a nucleus breeding program, it is possible to select replacement stock directly from the rearing piggery based on accuracy of GS used in the pre-selection strategy. The expected change in genetic gain from such a GS approach will be 173% and 38% greater returns compared to phenotypic and pedigree BLUP method, respectively, in selected population in high LD (Table 4). For unselected population in low LD, a greater return of 50% and 2% can be expected when GS is applied based on pre-selection accuracy compared to phenotypic and pedigree methods, respectively. In addition, the possibility of re-estimating marker allele substitution effects based only on small data set from the test station will lead to an expected reduction in annual returns of 15% and 5% for selected and unselected populations, respectively. Because recombination events can change marker-QTL phase configuration leading to a reduction in accuracy of GS and consequently, a reduction in the expected annual returns, marker effects should be re-estimated with large datasets (>2000 animals) whenever possible and after two generations of selection (Muir 2007).
|
Table 4. Expected genetic change per year for different selection schemes and populations |
|||||
|
Populations |
Methods |
ΔG/pig/year ($) 1 |
Return ($) |
Δ (%) 2 |
Δ (%) 3 |
|
Selected |
Phenotypic |
48 |
237,705 |
132 |
173 |
|
|
Pedigree |
94 |
469,147 |
17 |
38 |
|
|
Genomics |
110 |
550,470 |
- |
- |
|
|
PreGBV |
130 |
647,700 |
- |
- |
|
Unselected |
Phenotypic |
124 |
617,576 |
43 |
50 |
|
|
Pedigree |
182 |
909,861 |
-3 |
2 |
|
|
Genomics |
176 |
881,428 |
- |
- |
|
|
PreGBV |
186 |
928,055 |
- |
- |
|
15% of the males and 50% of the females were assumed to be selected annually 2Percent change in annual returns due to the use of GS based on marker effects re-estimation (Genomics) 3Percent change in annual returns due to the use of GS based on pre-selection accuracy (PreGBV) |
|||||
In the study by Schaeffer (2006), annual genetic gain was doubled because of a substantial reduction of generation intervals in three of the four pathways of selection in dairy cattle. Reduction of generation interval might be unfeasible with swine but by increasing the accuracy of genomic indices through the use of denser marker panel, annual genetic gain can be increased and this will offset the cost of genotyping animals.
By taking into account the cost of genotyping service and performance testing, the differential returns from various selection methods in a nucleus breeding program were compared (Tables 5 and 6). Depending on the selection method used, the pre-selection of 1000 pigs that entered the test station out of 5000 pigs produced annually in the nucleus offered an opportunity to reduce the cost of performance testing. The remaining 4000 pigs were expected to be sold to multipliers as additional returns. However, because accuracy of pre-selection differed with the method applied, it is possible to choose different breeding candidates to be performance tested by these methods. Consequently, good breeding candidates may be sent to the market rather than been fully performance tested. This is why it is important to establish an accurate method for pre-selection. Genome-wide selection offers an opportunity to have accurate GBV early in life of selection candidates and this will have an impact on expected rate of annual returns.
The total cost involved in the GS scheme when marker effects were re-estimated consisted of the cost of genotyping 5000 pigs in order to obtain GBV for pre-selection and the cost of performance testing 1000 pre-selected candidates. Starting at the current cost of genotyping for 60 K SNP panel which is around $120 per sample at DNA Landmarks (M. McNairnay, personal communication, 2011), several genotyping cost were evaluated by dropping the price by $30 up to a minimum price of $15 per sample. This provides an opportunity to find the optimum genotyping cost that will guarantee faster return per dollar invested when using GS at the current marker density of 60 K SNP chip. Cost of genotyping for thousands of marker is known to have reduced by a rate of 33% over a three year period (Simianer 2009). In this study prior estimates of marker effects were assumed available at no extra cost but in reality there is a cost. In the scenario where GS was applied based on accuracy of pre-selection strategy, cost of performance testing was ignored.
|
Table 5. Differential returns from different selection methods in a nucleus breeding program |
||||||
|
Populations |
Methods |
Genotyping cost/sample ($) |
Total returns ($) |
Total costs ($)1 |
Return on investment ($) |
Return per dollar invested ($) |
|
Selected |
Phenotypic |
0 |
237,705 |
55,000 |
182,705 |
3.32 |
|
|
Pedigree |
0 |
469,147 |
55,000 |
414,147 |
7.53 |
|
|
Genomics |
15 |
550,470 |
130,000 |
420,470 |
3.23 |
|
|
Genomics |
30 |
550,470 |
205,000 |
345,470 |
1.69 |
|
|
Genomics |
60 |
550,470 |
355,000 |
195,470 |
0.55 |
|
|
Genomics |
90 |
550,470 |
505,000 |
45,470 |
0.09 |
|
|
Genomics |
120 |
550,470 |
655,000 |
-104,529 |
-0.16 |
|
Unselected |
Phenotypic |
0 |
617,576 |
55,000 |
562,576 |
10.23 |
|
|
Pedigree |
0 |
909,861 |
55,000 |
854,861 |
15.54 |
|
|
Genomics |
15 |
881,428 |
130,000 |
751,428 |
5.78 |
|
|
Genomics |
30 |
881,428 |
205,000 |
676,428 |
3.30 |
|
|
Genomics |
60 |
881,428 |
355,000 |
526,428 |
1.48 |
|
|
Genomics |
90 |
881,428 |
505,000 |
376,428 |
0.75 |
|
|
Genomics |
120 |
881,428 |
655,000 |
226,428 |
0.35 |
|
1Total cost for GS scheme includes cost of performance testing 1000 candidates |
||||||
The total cost involved in the phenotypic and pedigree-based methods was only the cost of performance testing valued at about $55 per pig for 1000 tested candidates based on prices from a Canadian test station (B. Sullivan, personal communication, 2011). For the pedigree-based method, it was assumed that the infrastructure for implementing this method already existed. In tropical developing countries, where the infrastructure for swine genetic improvement using the conventional BLUP method are lacking, instituting a pedigree recording system for this environment will be a source of additional cost which can be very substantial.
The results presented in Table 5 suggests that if genotyping cost for 60 K SNP chip currently at $120 per sample can be reduced to $15 per sample, GS scheme will generate similar returns per dollar invested compared to phenotypic selection in a nucleus breeding program that uses a previously selected population in high LD. For the unselected population in low LD, GS method achieved a lower return than phenotypic selection at the same genotyping cost of $15 per sample. This suggests the importance of having LD in the population in order to apply GS. Also for all range of genotyping costs per sample, the GS scheme had lower returns per dollar invested compared to the pedigree BLUP method for the selected and unselected populations, respectively.
In the scenario where GS was applied based on accuracy of pre-selection strategy (Table 6), reduction of genotyping cost to $15 per sample will lead to an increase of $4.32 and 11 cents per dollar invested compared to phenotypic and pedigree BLUP methods, respectively, in previously selected population. At a genotyping cost of $30 per sample, GS can generate similar expected returns per dollar invested compared to currently practiced phenotypic selection in previously selected population. For unselected population, genotyping cost will need to be reduced to $15 per sample for the expected returns from GS to be greater than phenotypic selection by $1.14. Presently, the cost of genotyping can be reduced by genotype imputation (Howie et al 2009; Sargolzaei et al 2011). In a recent study in dairy cattle, Sargolzaei et al (2011) imputed 50 K genotypes from 6 K SNP panel with accuracy of about 99%. The cost of genotyping an animal for the 6 K SNP panel is around $40 at DNA Landmarks (M. Sargolzaei, personal communication, 2011). This technology can be applied to swine breeding, thus reducing the genotyping cost.
|
Table 6. Differential returns from selection when GS scheme is based on pre-selection accuracy in a nucleus breeding program |
||||||
|
Populations |
Methods |
Genotyping cost/sample ($) |
Total returns ($) |
Total costs ($)1 |
Return on investment ($) |
Return per dollar invested ($) |
|
Selected |
Phenotypic |
0 |
237,705 |
55,000 |
182,705 |
3.32 |
|
|
Pedigree |
0 |
469,147 |
55,000 |
414,147 |
7.53 |
|
|
PreGBV |
15 |
647,700 |
75,000 |
572,700 |
7.64 |
|
|
PreGBV |
30 |
647,700 |
150,000 |
497,700 |
3.32 |
|
|
PreGBV |
60 |
647,700 |
300,000 |
347,700 |
1.16 |
|
|
PreGBV |
90 |
647,700 |
450,000 |
197,700 |
0.49 |
|
|
PreGBV |
120 |
647,700 |
600,000 |
47,700 |
0.08 |
|
Unselected |
Phenotypic |
0 |
617,576 |
55,000 |
562,576 |
10.23 |
|
|
Pedigree |
0 |
909,861 |
55,000 |
854,861 |
15.54 |
|
|
PreGBV |
15 |
928,055 |
75,000 |
853,055 |
11.37 |
|
|
PreGBV |
30 |
928,055 |
150,000 |
778,055 |
5.19 |
|
|
PreGBV |
60 |
928,055 |
300,000 |
628,055 |
2.09 |
|
|
PreGBV |
90 |
928,055 |
450,000 |
478,055 |
1.06 |
|
|
PreGBV |
120 |
928,055 |
600,000 |
328,055 |
0.55 |
|
1Total cost for GS scheme excludes cost of performance testing |
||||||
A major concern while genome-wide selection is conjectured to be disadvantaged compared to conventional approach is the cost of genotyping animals for millions of marker loci. As genotyping cost decreases and marker density that is greater than 60 K SNP panel assumed in the current study become available in future, QTL allele effects are expected to be estimated with more precision due to higher LD and leading to greater accuracy of selection and greater returns per dollar invested. The overall results from this study indicates that, although the returns per dollar invested are greater for conventional BLUP selection method ignoring cost of instituting a pedigree recording system for developing countries, the marginal rates of returns to selection using GS are expected to be greater as cost of genotyping for 60 K SNP panel become reduced to $30 per sample or as next generation sequencing technology become feasible for swine breeding in future.
Relative economic returns from the implementation of genome-wide selection in a nucleus swine breeding program are possible. Benefits arise from an increase in accuracy of genomic indices associated with a possible substantial reduction in the cost of genotyping for 60 K SNP panel to $30 per sample in the future and from the potential elimination of performance testing cost for a population in high LD compared to phenotypic selection that is currently used in developing countries. For unselected populations in low LD, denser marker panels will need to be used in order to increase accuracy of selection. In addition, individuals entering a performance test station in a nucleus breeding program can be pre-selected with greater accuracy by using a genomic index compared to selection indices that are based on parent average. However, will farmers accept genomic indices as a selection criterion without having a prior knowledge of the performance of selection candidates in a test station? This will be the crucial point for practical implementation of GS in a nucleus swine breeding program in future.
The first author acknowledges funding from the Canadian Commonwealth Scholarship program under the Department of Foreign Affairs and International Trade (DFAIT).
Akanno E C, Schenkel F S, Quinton V M, Friendship R M and Robinson J A B 2013 Meta-analysis of genetic parameter estimates for reproduction, growth and carcass traits of pigs in the tropics. Livestock Science, http://dx.doi.org/10.1016/j.livsci.2012.07.021
Aker C and Kennedy B W 1988 Estimated Breeding Value: A tool for genetic improvement of swine. In: FACTSHEET. Ontario ministry of agriculture, food and rural affairs, order number 88 – 014
Amaral A J, Hendrik-Jan M, Crooijmans R P M A, Heuven H C M and Groenen M A M 2008 Linkage disequilibrium decay and haplotype block structure in pig. Genetics 179: 569-579
Bulmer M 1976 The effect of selection on genetic variability: A simulation study. Genetics Research 28(02):101-117
de Roo G 1987 A stochastic model to study breeding schemes in a small pig population. Agricultural Systems 25(1):1-25
Dekkers J C M 2007 Prediction of response to marker-assisted and genomic selection using selection index theory. Journal of Animal Breeding and Genetics 124:331-341
Du F X, Clutter A C and Lohuis M M 2007 Characterizing linkage disequilibrium in pig populations. International Journal of Biological Sciences 3(3):166-178
Dzama K 2002 The pig performance testing scheme in Zimbabwe. AGTR Case study,Nairobi, Kenya, ILRI
Fernando R L, Habier D, Stricker C, Dekkers J C M and Totir L R 2007 Genomic selection. Acta Agriculturae Scandinavica, Section A – Animal Sciences 57(4): 192-195
Garrick D J and van Vleck L D 1987 Aspects of selection for performance in several environments with heterogeneous variances. Journal of Animal Science 65:409-421
Gilmour A R, Gogel B J, Cullis B R and Thompson R 2009 ASReml user guide release 3.0.3
Habier D, Fernando R L and Dekkers J C M 2007 The impact of genetic relationship information on genome-assisted breeding values. Genetics 177(4): 2389-2397
Hayes B and Goddard M E 2001 The distribution of the effects of genes affecting quantitative traits in livestock. Genetics Selection Evolution 33(3):209-229
Henderson C R 1984 Applications of linear models in animal breeding. University of Guelph, Guelph, Canada.
Howie B N, Donnelly P and Marchini J 2009 A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genetics 5(6):e1000529 doi:10.1371/journal.pgen.1000529
Ihaka R and Gentleman R 1996 R: A language for data analysis and graphics. Journal of Computational and Graphical Statistics 5(3):299-314
Kahi A K, Rewe T O and Kosgey I S 2005 Sustainable community-based organizations for the genetic improvement of livestock in developing countries. Outlook Agriculture 34(4):261-270
König S and Swalve H H 2009 Application of selection index calculations to determine selection strategy in genomic breeding programs. Journal of Dairy Science 92:5292-5303
Kuhn M T, Boettcher P J and Freeman A E 1994 Potential bias in transmitting abilities of females from preferential treatment. Journal of Dairy Science 77:2428-2437
Meuwissen T H E, Hayes B J and Goddard M E 2001 Prediction of total genetic value using genome-wide dense marker maps. Genetics 157(4):1819-1829
Muir W M 2007 Comparison of genomic and traditional BLUP - estimated breeding value accuracy and selection response under alternative trait and genomic parameters. Journal of Animal Breeding and Genetics 124(6):342-355
Ramos A M, Crooijimans R P, Affara N A, Amaral A J, Archibald A L, Beever J E, Bendixen C, Churcher C, Clark R, Dehais P, Hansen M S, Hedegaard J, Hu Z L, Kerstens H H, Law A S, Megens H J, Milan D, Nonneman D J, Rohrer G A, Rothschild M F, Smith T P, Schnabel R D, Van Tassell C P, Taylor J F, Wiedmann R T, Shook L B and Groenen M A 2009 Design of high density SNP genotyping assay in the using of SNPs identified and characterized by next generation sequencing technology. PLoS One 4:e6524
Sargolzaei M, Schenkel F and Chesnais J 2011 Accuracy of imputed 50k genotypes from 3k and 6k chips using FImpute version 2. Technical report to the Dairy Cattle Breeding and Genetics Committee Meeting, September 13, 2011
Schaeffer L R 2006 Strategy for applying genome-wide selection in dairy cattle. Journal of Animal Breeding and Genetics 123(4):218-223
Simianer H 2009 The potential of genomic selection to improve litter size in pig breeding programs. Proceedings of the 60th Annual Meeting of European Association of Animal Production, Barcelona, Spain
Sitzenstock F, König S, Ytournel F and Simianer H 2010 Evaluation of genomic selection for functional traits in horse breeding programs. Proceedings of the 9th World Congress on Genetics Applied to Livestock Production, Leipzig, Germany
Smith C 1988 Genetic improvement of livestock in developing countries using nucleus breeding units. World Animal Review. 65:2–10
Received 24 November 2012; Accepted 15 February 2013; Published 1 March 2013